Single-Cell vs. Spatial: When to Use Which (and When to Use Both)
A decision framework for choosing between single-cell RNA-seq and spatial transcriptomics for your study — based on the biological question, not the technology hype.
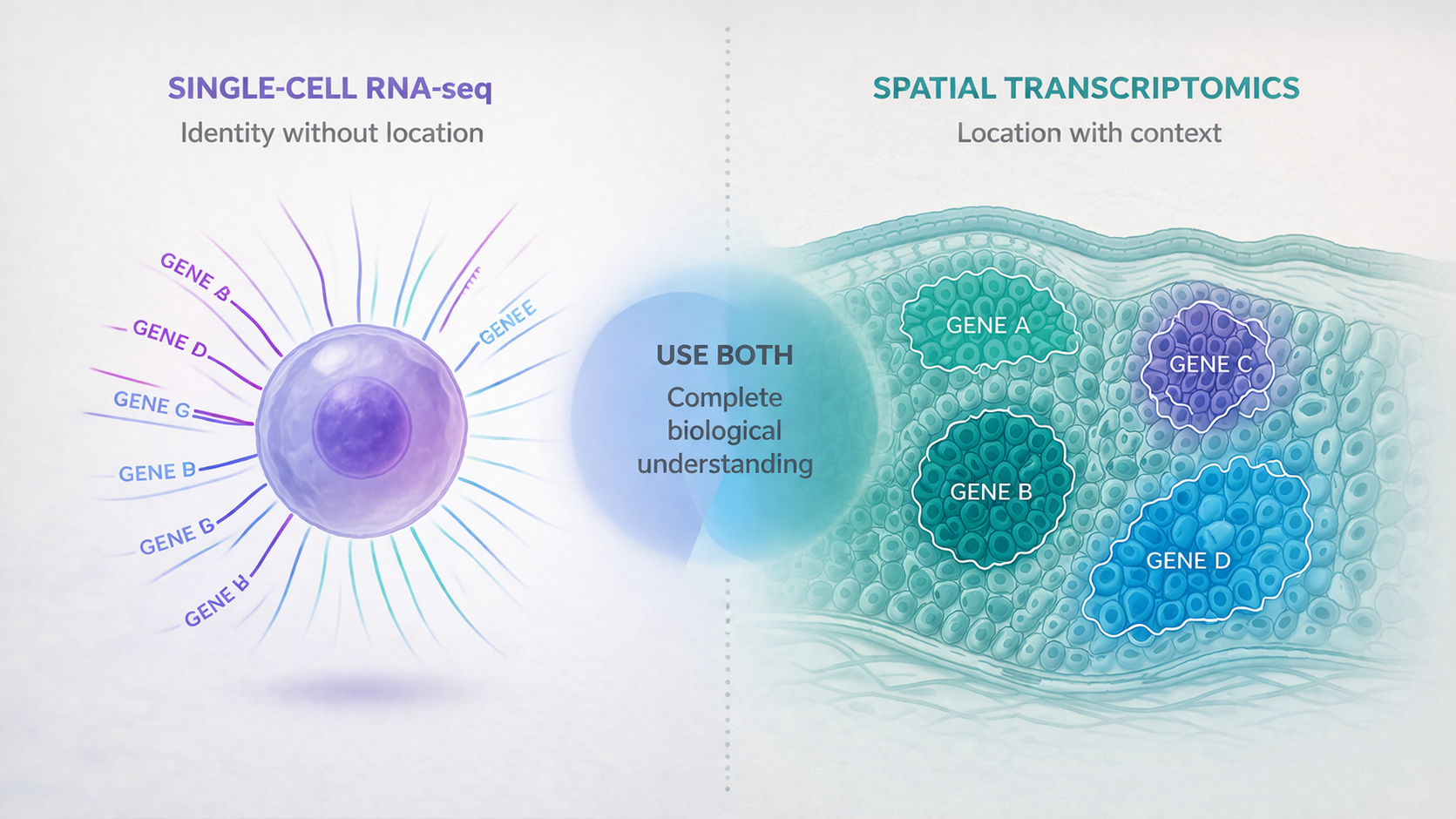
“Should I do single-cell or spatial?”
This is one of the most common questions we get from researchers planning new studies. The answer is almost never “one or the other” — it depends on what you’re trying to learn. The real difference isn’t resolution or technology — it’s whether you need cellular identity or spatial context. The decision framework is simpler than most people think.
What Each Technology Tells You
Single-Cell RNA-seq (scRNA-seq)
Single-cell RNA-seq dissociates tissue into individual cells, captures each cell’s transcriptome, and sequences them. You get:
- Cell identity: What cell types are present, in what proportions
- Cellular heterogeneity: Subpopulations within a cell type (e.g., exhausted vs. effector T cells)
- Rare cell detection: Populations representing <1% of the sample (depending on sampling depth)
- Trajectory analysis: Developmental or differentiation pathways
- Gene regulatory networks: Co-expression patterns at single-cell resolution
What you lose: where those cells were in the tissue. Dissociation destroys spatial context.
In short, scRNA-seq excels at defining what cells are present and what they are doing.
Spatial Transcriptomics
Spatial transcriptomics profiles gene expression while preserving tissue architecture. Depending on the platform, you get:
- Location: Where each cell (or region) sits within the tissue
- Neighborhoods: What cells are adjacent to each other
- Compartment-specific biology: Tumor vs. stroma vs. immune regions analyzed separately
- Tissue architecture: How gene expression relates to histological features
- In situ biology: No dissociation artifacts
There is a fundamental tradeoff in spatial technologies: you typically choose between whole-transcriptome coverage (GeoMx, Visium) and single-cell resolution (CosMx, MERFISH), but rarely get both at full depth simultaneously.
Spatial transcriptomics answers where biology happens — and how cells interact within tissue context.
The Decision Framework
Choose scRNA-seq when:
1. You need to discover unknown cell types or states.
If you don’t know what cell populations exist in your tissue, scRNA-seq is the discovery tool. Unsupervised clustering will reveal populations you didn’t anticipate — and many of the most important discoveries in single-cell biology have been unexpected cell states.
2. You need rare cell detection.
Spatial platforms have detection limits. A cell type representing 0.5% of the tissue is difficult to identify with ROI-based methods (GeoMx) and may be missed by spot-based methods (Visium). scRNA-seq can detect these populations if you sequence enough cells.
3. You’re studying dissociated systems.
Blood, bone marrow, PBMC, and cell culture experiments don’t have meaningful spatial context. Single-cell is the natural choice for these sample types.
4. You need trajectory or pseudotime analysis.
Developmental trajectories, differentiation pathways, and cell-state transitions are best resolved with scRNA-seq tools like Monocle3, scVelo, and RNA velocity.
Choose spatial transcriptomics when:
1. Location matters.
If your question is about where gene expression happens — at the tumor margin, in the stroma, adjacent to blood vessels — you need spatial resolution. scRNA-seq destroys this information.
2. You’re studying tissue microenvironments.
Cell-cell interactions, immune exclusion, niche biology, and compartment-specific gene expression all require spatial context. The tumor-immune interface, for example, is a spatial concept that cannot be reconstructed from dissociated cells.
3. You have FFPE tissue only.
Many clinical and archival samples are only available as FFPE blocks. If your only material is archival FFPE, spatial platforms are often the most practical option. Nuclei-based methods like snRNA-seq exist and FFPE-compatible single-cell approaches are emerging, but they are not equivalent for all applications. Spatial platforms (GeoMx, CosMx, Visium HD) work with FFPE.
4. You need RNA + protein from the same section.
Multi-omic spatial profiling (GeoMx, CosMx) provides matched RNA and protein measurements from the same tissue region — something scRNA-seq cannot offer.
5. Dissociation artifacts are a concern.
Enzymatic dissociation changes gene expression. Stress response genes (FOS, JUN, heat shock proteins) are upregulated during dissociation, and some cell types (neurons, adipocytes, large cells) are lost entirely. Spatial profiling avoids these artifacts.
Use both when:
1. You want to deconvolve spatial data with single-cell resolution.
The most powerful combination: use scRNA-seq to define cell type signatures, then use those signatures as the reference matrix for spatial deconvolution. This gives you cell-type-resolved spatial information without requiring single-cell spatial platforms.
2. You’re building a comprehensive tissue atlas.
Large-scale tissue mapping projects (like the Human Cell Atlas) combine scRNA-seq for cell type discovery with spatial methods for tissue mapping. Each technology validates and extends the other.
3. You need to validate spatial findings at single-cell resolution.
A spatial observation (e.g., “this tissue compartment is enriched for a specific pathway”) can be validated and deepened by scRNA-seq of the same tissue, confirming which cell types drive the signal.
4. Budget and sample availability allow it.
If you have fresh tissue for scRNA-seq and adjacent sections for spatial profiling, doing both provides a richer dataset than either alone.
One important caveat: even with perfect scRNA-seq data, inferred spatial methods (like imputing spatial coordinates from expression) are approximations, not replacements for direct spatial measurement. These inference methods are improving rapidly, but if spatial context is central to your question, direct spatial profiling remains the most reliable approach.
In practice, the choice is often less about which technology is “better” and more about which information would be impossible to recover later if you didn’t collect it now. If you dissociate tissue for scRNA-seq, the spatial context is gone forever. If you profile spatially but at regional resolution, the single-cell heterogeneity within those regions is lost. Thinking about irreversibility often clarifies the decision.
Common Mistakes
Choosing spatial because it’s newer
Spatial transcriptomics is exciting, but if your question doesn’t require spatial context, scRNA-seq may be simpler, cheaper, and more informative. Don’t choose a platform because it’s trendy.
Choosing scRNA-seq because it’s familiar
Conversely, many research groups default to scRNA-seq because they have the computational pipelines in place. If your biological question is fundamentally spatial (immune exclusion, tissue architecture, compartment-specific biology), scRNA-seq will miss the point no matter how well you analyze it.
Underestimating the computational burden
Both technologies require significant bioinformatics support, but the nature of the support differs:
- scRNA-seq: Cell filtering, normalization, batch correction, clustering, annotation, trajectory analysis. Well-established tools (Seurat, Scanpy) but requires expertise.
- Spatial: ROI selection strategy (GeoMx), spot deconvolution (Visium), segmentation (CosMx/MERFISH), compartment-specific analysis, spatial statistics. Newer tools, fewer established best practices.
Ignoring sample size
Both technologies are expensive per sample. Plan your study design to ensure adequate statistical power:
- scRNA-seq: Often requires multiple biological replicates per condition (commonly 3-5+, depending on study design), with several thousand cells per sample
- Spatial: Varies by platform and study design — GeoMx typically needs multiple ROIs per compartment per condition; Visium needs multiple tissue sections
A Practical Decision Checklist
Answer these questions to guide your choice:
- Does my question require knowing where gene expression happens in tissue? → Spatial
- Do I need to discover unknown cell populations? → scRNA-seq
- Is my only material archival FFPE? → Spatial is often the most practical choice
- Do I need RNA + protein together? → Spatial (GeoMx, CosMx)
- Am I studying a dissociated system (blood, cell culture)? → scRNA-seq
- Do I need rare cell detection (<1%)? → scRNA-seq
- Am I concerned about dissociation artifacts? → Spatial
- Do I want cell-type-resolved spatial information? → Both (scRNA-seq for signatures + spatial for mapping)
How We Help
At Cytogence, we support both single-cell and spatial transcriptomics projects — and we help researchers make the platform decision before they commit experimental resources. Choosing the wrong platform is one of the most expensive mistakes in experimental design. If you’re planning a study and need guidance on which approach (or combination) will best answer your biological question, we’d like to help you think it through.
Cytogence provides bioinformatics consulting for single-cell RNA-seq, spatial transcriptomics, and multi-omics research. Contact us to discuss your study design.